.jpg)
Warum das wichtig ist: Cystische Fibrose, oft auch Mucoviscidose genannt, ist die häufigste genetische Stoffwechselstörung in Westeuropa.
4 Prozent aller Westeuropäer:innen tragen eine genetische Mutation in sich, die sie an ihre Kinder weitergeben können, sodass diese erkranken (USZ, 2022)
In Österreich kommt etwa alle zwei Wochen ein krankes Kind zur Welt. (CF austria, 2022). Dank des Frühgeborenen-Screenings erkennt man diese Erkrankung bereits bei der Geburt, doch die Behandlung gestaltet sich schwierig. Marcus Mall, ein Kinderfacharzt aus Berlin, arbeitet seit vielen Jahren an einer Methode, diese Krankheit zu heilen. Für die Entwicklung einer neuen Therapie wurde er heuer beim Falling Walls Science Summit in Berlin ausgezeichnet.
Evas Geschichte
„Es ist mir eine große Freude, dass Eva heut hier im Publikum ist. Ich möchte sie auf die Bühne einladen.“ Applaus bricht los, während eine junge Frau mit kurzem blondem Bob in einem schwarzen Kleid die Bühne betritt. Für eine Minute übertönt tosender Applaus jedes andere Geräusch.
Eva leidet an Cystischer Fibrose, an jener Erkrankung, die Marcus Mall zu heilen versucht. In einem Video hat der Arzt von der Geschichte seiner Patientin erzählt. Nun steht sie neben ihm auf der Bühne und berichtet von den Problemen, die sie jeden Tag begleiten. Das Leid, das ihre Krankheit verursacht, macht sie für Wissenschaftler:innen aus aller Welt sichtbar. Plötzlich handelt es sich nicht mehr um eine gesichtslose Erkrankung mit einem komplizierten Namen – es handelt sich um Schicksale. Und um ein Problem, das behandelt werden muss.
Was ist Cystische Fibrose?
Doch welches Problem stellt Cystische Fibrose genau dar? Das hat der Forscher bereits zu Beginn seines Vortrages den Anwesenden erklärt: „Patient:innen beschreiben es so: Sie fühlen sich, als wäre ihre Lunge stets mit Wasser gefühlt, sodass das Atmen beinahe unmöglich wird.“
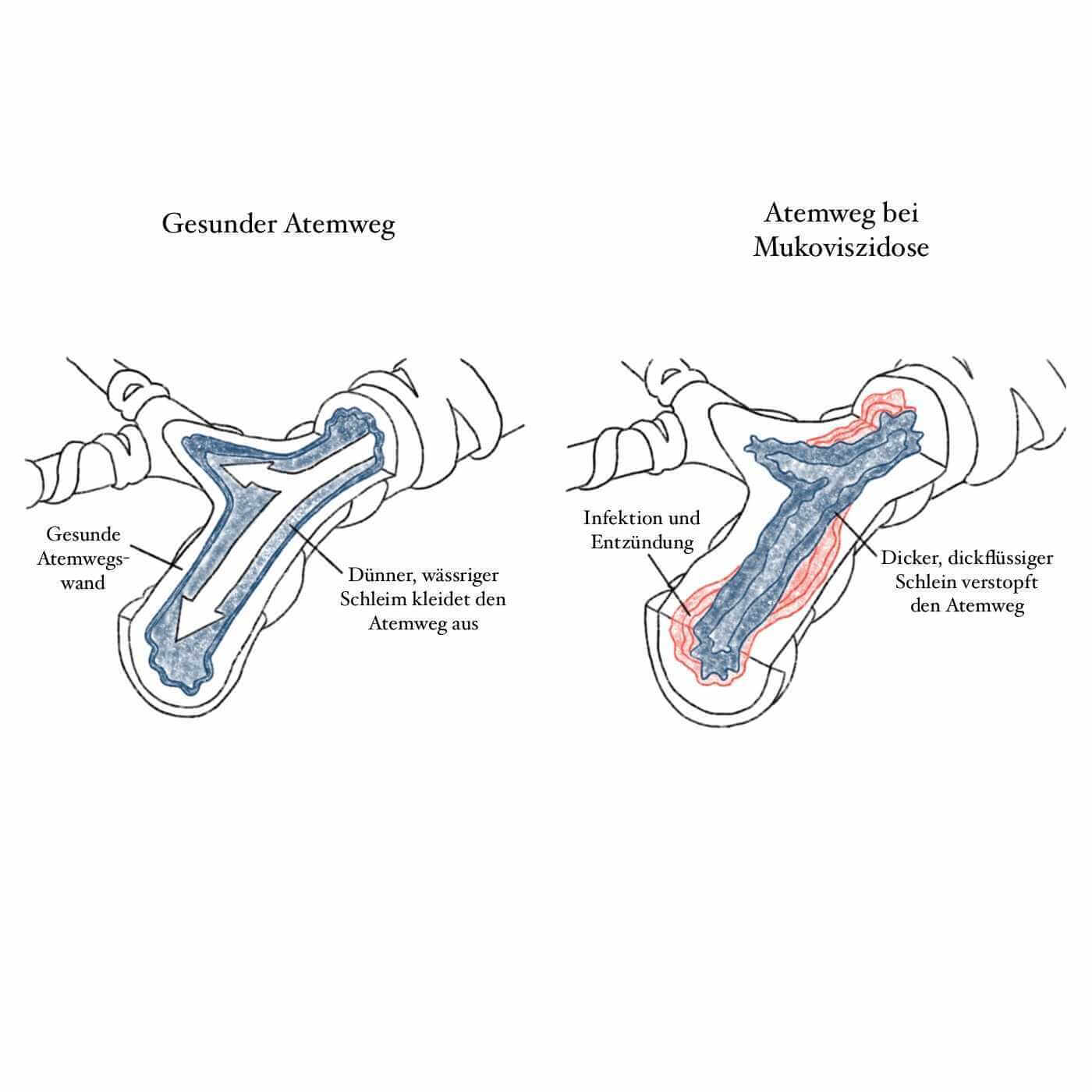
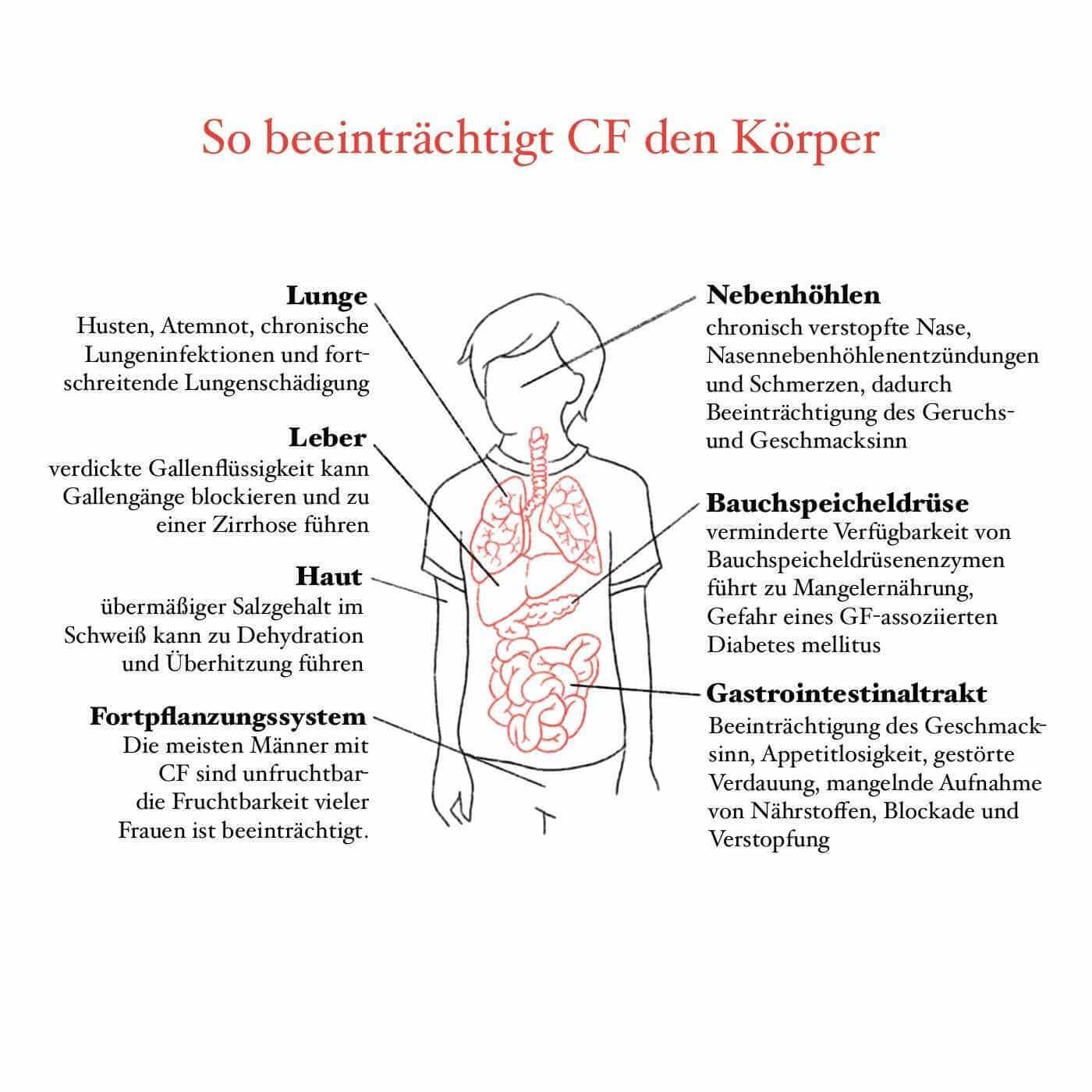
Bedingt ist dieses Gefühl durch einen zähen Schleim (auch Mukus genannt), der die Atemwege der Betroffenen auskleidet, sodass die Luft nicht frei strömen kann. Das allein führt bereits zu einer massiven Abnahme der Lungenfunktion. Menschen, die an Cystischer Fibrose erkrankt sind, haben im besten Fall nur die Hälfte der Lungenfunktion eines Menschen ohne Gendefekt.
Zusätzlich bietet der zähe Schleim eine perfekte Grundlage für Bakterien, sodass die Patient:innen regelmäßig an Infektionen leiden und immer wieder mit Entzündungen zu kämpfen haben. Diese chronischen Entzündungen zerstören nach und nach das Gewebe, sodass es irgendwann zu irreversiblen Lungenschädigungen kommt. Daher benötigen die meisten Patient:innen irgendwann eine Lungentransplantation.
Das erklärt die verkürzte Lebenserwartung der Erkrankten, die auch mit den Therapien, die in den letzten Jahren entwickelt wurden, maximal 50 Jahre beträgt. Noch dazu geht diese kurze Lebenserwartung mit einer geringen Lebensqualität einher. Die Betroffenen müssen stundenlang inhalieren, um den Schleim ein wenig zu lösen, und bis zu dreißig Medikamente pro Tag zu sich nehmen.
Denn nicht nur die Lungen, sondern auch andere Organe sind von dieser Erkrankung betroffen – es handelt sich bei Cystischer Fibrose um eine sogenannte Multisystemerkrankung. So kann die Bauchspeicheldrüse aufgrund des zählen Schleimes ihre Verdauungsenzyme nicht in den Verdauungstrakt abgeben. Es kommt zu einer sogenannten exokrinen (von Drüsen ausgehend) Pankreasinsuffizienz.
Da dadurch die Ernährung nicht vollständig verdaut werden kann, leiden Kinder oft an Gedeihstörungen, das bedeutet, es kommt zu Wachstumsverzögerungen und zu einer mangelnden Gewichtszunahme. Zusätzlich ist der Schweiß von Patient:innen mit Cystischer Fibrose wesentlich salzreicher, da auch dessen Produktion nicht gut funktioniert. Um diesen starken Salzverlust und den damit drohenden Salzmangel auszugleichen, müssen Patient:innen besonders viel Salz zu sich nehmen.
Eva erzählt uns, dass sie früher ständig außer Atem und immer müde war. Aufgrund ihrer Erkrankung musste sie vor einigen Jahren ihre Arbeit aufgeben. Außerdem hatte sie nie die Kraft, ihren dreijährigen Sohn zu tragen, geschweige denn mit ihm zu spielen.
Um dies zu unterstreichen, zeigt Marcus Mall ein kurzes Video, das einen kleinen Jungen beim Laufen zeigt. Bald schon bleibt er stehen, als seine Mutter ihm hinterherruft: „Edgar, nicht so schnell, Mama kann nicht so schnell.“ Das ist ein Schmerz, der bei keiner Untersuchung festgestellt werden kann, und der dennoch das Leben von Eva durchzieht.
Das CFTR-Gen und seine Mutationen
Die genetische Erkrankung wurde erstmals 1930 entdeckt, die genaue Ursache war damals unbekannt. Die meisten Kinder, die mit dieser Krankheit zur Welt kamen, erreichten nicht einmal das Schulalter.
Der Krankheit liegt ein genetischer Defekt im CFTR-Gen zugrunde, der erst 1989 als Verursacher der Erkrankung identifiziert werden konnte. Seit 1998 ist bekannt, welche Mutationen das Gen unbrauchbar machen. Seither versuchten Wissenschaftler:innen aus aller Welt diesen Gendefekt zu beheben – mittels Gentherapie. Doch bisher erwiesen sich die Versuche als Fehlschläge.
Bei 90 Prozent der bisher 1.000 identifizieren Mutationen, die das CFTR-Gen unbrauchbar machen und Cystische Fibrose auslösen, handelt es sich um die sogenannte F508del-Mutation. Dadurch kommt es zu einer Fehlfaltung des gebildeten Proteins, sodass es gleich nach der Fertigstellung von unseren Zellen wieder abgebaut wird. Dadurch wird der wesentliche Chlorid-Kanal nicht in die Zellmembran eingebaut. Ohne diesen Chlorid-Kanal können jedoch Drüsen keinen gut transportierbaren, wässrigen Schleim produzieren und ein zäher Schleim bleibt zurück.
Eine Triple-Therapie als Heilung
Marcus Mall, der bereits 1998 an der Entdeckung der F508 del-Mutation beteiligt war, forschte den Großteil seiner Karriere an einer Lösung dieses Problems. Mit seinem Team gelang es ihm in den letzten Jahren Modulatoren zu entwickeln, um die Fehlfaltung des Proteins aufzuheben und so funktionstüchtig zu machen.
Im Jahre 2021 veröffentlichte das Team rund um Marcus Mall eine Phase III-Studie, die drei Wirkstoffe kombiniert. Zwei davon, Tezacaftor und Elaxacaftor, sorgen dafür, dass die Fehlfaltung des Proteins aufgehoben wird und verbessern so die Lungenfunktion.
Ivacaftor, der dritte Wirkstoff dieser Therapie, verbessert den Einbau des Proteins in die Zellmembran, wo es dann als Chlorid-Kanal fungiert. Zusammen steigern sie die Lungenfunktion und vermindern die Häufigkeit von Lungenentzündungen. Unter dieser Therapie konnte die Lungenfunktion der Proband:innen der Studie beinahe um das doppelte verbessert werden, also beinahe auf das Niveau einer gesunden Person. Zusätzlich zu diesen molekularen Veränderungen erhöht sich auch die Lebensqualität der Patient:innen erheblich.
Das bestätigt uns Eva. Bereits nach vier Wochen hatte sie viel mehr Energie. Mittlerweile benötigt sie nur noch die Hälfte aller Medikamente, die sie vor der neuen Therapie einnehmen musste. Sie hat wieder zu arbeiten begonnen und ist einige Stunden pro Tag in ihrem Beruf tätig.
Das Wichtigste für sie: Sie kann ihren Sohn mittlerweile tragen. Und sie besitzt genug Kraft, um ihn durch sein Leben zu begleiten. Während Marcus Mall und sie das nächste kurze Video zeigen, in dem sie ihrem Sohn auf seinem Rad hinterherläuft, kämpft Eva mit den Tränen. Auch das Publikum ist sichtlich bewegt.
Keine Wunder ohne Opfer
So erfolgreich und vielversprechend die Therapie von Marcus Mall auch ist, es gibt keine Medikation, die keinerlei Nebenwirkungen verursacht. Das betont auch der Arzt selbst. Derzeit sind noch keine Langzeit-Nebenwirkungen bekannt, dafür wird das Medikament noch nicht lange genug eingesetzt. In den letzten fünf Jahren konnten jedoch bereits einige Nebenwirkungen vermerkt werden.
So kommt es bei einigen Patient:innen zu Erhöhung der Leberwerte im Blut, also einer möglichen Leberschädigung, die jedoch meist von selbst wieder verschwinden. Nur bei einigen wenigen Patient:innen musste deswegen eine Therapiepause oder sogar ein Absetzen der Therapie erfolgen.
Allerdings muss man hinzufügen, dass es kaum ein Medikament gibt, das nicht entweder Leber oder Niere beeinträchtigt. Diese beiden Organe haben nämlich die Aufgabe, sämtliche Stoffe, die wir unserem Organismus zuführen, wieder abzubauen. Das führt eben oftmals zu einem möglichen Zellverbrauch. Menschen, die aufgrund einer Erkrankung regelmäßig und viele Medikamente einnehmen müssen, können so ihre Leber und Niere strapazieren.
Eine weitere Nebenwirkung ist eine starke Gewichtszunahme. Dies kommt daher, dass Kinder und Erwachsene, die an Cystischer Fibrose leiden, mehr Kalorien (vor allem Fett) zu sich nehmen müssen als gesunde Menschen, da sie einen Großteil nicht verdauen können. Unter der neuen Therapie konnte das Team von Marcus Mall feststellen, dass viele Patient:innen plötzlich übergewichtig werden, sogar stark übergewichtig, was gesundheitliche Folgen hat.
Der Grund: Durch die Therapie verbessert sich die Bauchspeicheldrüsenfunktion. Das führt jedoch dazu, dass die bisher fettreiche und hochkalorische Ernährung komplett umgestellt werden muss, um die Gewichtszunahme zu vermeiden.
Da auch der Salzverlust über den Schweiß unter der neuen Medikation zurückgeht, sollten therapierte Patient:innen neben dem Kaloriengehalt auch das Salzgehalt in ihrer Ernährung reduzieren. Die bisherigen Studien zeigten, dass viele Betroffene im Laufe der Zeit einen Bluthochdruck entwickeln. Denn durch die nun zu hohe Salzzufuhr steigt der Blutdruck an.
Andere schwere Nebenwirkungen sind derzeit nicht bekannt, sodass nach Therapiebeginn vor allem die Ernährungsumstellung für Patient:innen wichtig ist. Ein Opfer, das sicher alle Patient:innen zu erbringen bereit sind, wenn eine Linderung ihrer Symptome und eine Verbesserung der Lebensqualität in greifbare Nähe rücken.
Wie es in Zukunft weitergehen soll
Derzeit ist diese Therapie für Kinder ab sechs Jahren zugelassen. Marcus Mall arbeitet daran, dass sie auch für jüngere Kinder möglich wird, am besten ab dem Tag der Geburt. Denn durch die irreversible Schädigung der Lunge ist es seiner Meinung nach wesentlich, Patient:innen die Therapie so früh wie möglich zukommen zu lassen.
Der Kinderarzt erwähnt, dass bereits eine weitere Studie, in der Kinder von zwei bis sechs Jahren behandelt werden, abgeschlossen wurde. Die Daten werden derzeit noch ausgewertet. Wenn auch die Ergebnisse dieser Studie eine erfolgreiche Therapie mit geringen Nebenwirkungen versprechen, stehen die Chancen gut, dass die amerikanische Arzneimittelbehörde FDA und die europäische Behörde EMA diese bald zulassen.
Der nächstlogische Schritt ist für den Arzt eine Studie, in welchem der Wirkstoff bei Neugeborenen getestet wird. So könnten in Zukunft 90 Prozent aller Personen, die von Cystischer Fibrose betroffen sind, ein beinahe normales Leben führen. Den restlichen 10 Prozent der Patient:innen, die an einer anderen Mutation leiden, soll natürlich auch geholfen werden. Daher forschen bereits einige Wissenschaftler:innen an Gentherapien, um in Zukunft allen Menschen, die an Cystischer Fibrose erkrankt sind, helfen zu können. Damit alle Mütter und Väter, die an Cystischer Fibrose leiden, genau wie Eva ihre Kinder hochheben und durch das Leben tragen können.
CF austria (2022). CF die Krankheit. Cystische Fibrose Hilfe Österreich.
USZ (2022). Zystische Fibrose. Universitätsspital Zürich.
Barry, P.J., Mall, M.A., Álvarez, A., et al. (2021). Triple Therapy for Cystic Fibrosis
Phe508del–Gating and –Residual Function Genotypes. N Engl J Med, 385(9):
815–825.
Middleton, P.G., Mall, M.A., Dřevínek, P., et al. (2019). Elexacaftor-Tezacaftor-Ivacaftor
for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med, 381(19): 1809-1819.


.png)